
Page 192 - 《精细化工》2020年第11期
P. 192
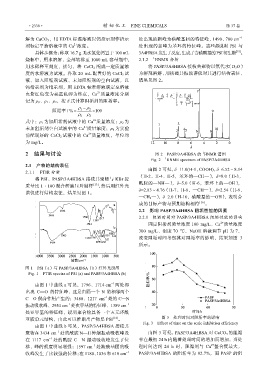
·2338· 精细化工 FINE CHEMICALS 第 37 卷
解为 CaCO 3 ,用 EDTA 标准溶液以钙指示剂作指示 处出现的新峰为磺酸基团的特征峰,1490、700 cm –1
2+
剂标定平衡溶液中的 Ca 浓度。 处出现的新峰为苯环的特征峰。这些都说明 PSI 与
[15]
具体步骤为:称取 16.7 g 无水氯化钙置于 100 mL 3A4HBSA 发生了反应,生成了含磺酸基的PSI 衍生物 。
1
烧杯中,用水溶解,全部转移至 1000 mL 容量瓶中, 2.1.2 HNMR 分析
用水稀释至刻度,摇匀,将 CaCl 2 配成一定质量浓 将 PASP/3A4HBSA 接枝共聚物以氘代水(D 2 O)
度的水溶液为试液。各取 20 mL 配置好的 CaCl 2 试 为溶剂溶解,用核磁共振波谱仪对其进行结构表征,
液、加入阻垢剂试液、未加阻垢剂的空白试液,以 结果见图 2。
钙指示剂为指示剂,用 EDTA 标准溶液滴定至溶液
2+
由紫红色变为亮蓝色即为终点,Ca 质量浓度分别
记为 ρ 0、ρ 1、ρ 2 ,按下式计算阻垢剂的阻垢率。
阻垢率 /% 1 2 100
0 2
2+
式中:ρ 1 为加阻垢剂试液中的 Ca 质量浓度;ρ 2 为
2+
未加阻垢剂空白试液中的 Ca 质量浓度;ρ 0 为实验
2+
前配制好的 CaCl 2 试液中的 Ca 质量浓度,单位均
为 mg/L。
2 结果与讨论 图 2 PASP/3A4HBSA 的 HNMR 谱图
1
1
Fig. 2 HNMR spectrum of PASP/3A4HBSA
2.1 产物的结构表征
由图 2 可见,δ=11.0(H-1,COOH),δ=6.52~8.14
2.1.1 FTIR 分析
(H-2、H-4、H-5,苯环的—CH—),δ=8.0(H-3,
将 PSI、PASP/3A4HBSA 接枝共聚物与 KBr 按
质量比 1∶100 混合研磨压片制样 [13] ,然后用红外光 酰胺的—NH—),δ=5.0(H-6,苯环上的—OH),
δ=2.83~4.76(H-7、H-8,—CH—),δ=2.54(H-9,
谱仪进行结构表征,结果见图 1。
—CH 2 —),δ=2.0(H-10,磺酸基的—OH),表明合
成的目标产物与预期结构相符 [16] 。
2.2 影响 PASP/3A4HBSA 阻垢性能的因素
2.2.1 阻垢时间对 PASP/3A4HBSA 阻垢性能的影响
2+
固定阻垢剂质量浓度 100 mg/L、Ca 质量浓度
300 mg/L、温度 70 ℃,NaOH 溶液调节 pH 为 7,
改变阻垢时间考察其对阻垢率的影响,结果如图 3
所示。
图 1 PSI(a)与 PASP/3A4HBSA(b)红外光谱图
Fig. 1 FTIR spectra of PSI (a) and PASP/3A4HBSA (b)
–1
由图 1 中曲线 a 可见,1796、1714 cm 两处都
出现 C==O 的特征峰,这是由隔一个 N 的相邻两个
–1
C==O 偶合作用产生的;3488、1217 cm 处的 C—N
–1
振动吸收峰、2954 cm 处次甲基的特征峰、1389 cm –1
处亚甲基的特征峰,说明聚合物具备一个五元环酰
亚胺单元结构,由此可以推断出产物是 PSI [14] 。 图 3 处理时间对阻垢率的影响
Fig. 3 Effect of time on the scale inhibition efficiency
由图 1 中曲线 b 可见,PASP/3A4HBSA 接枝共
–1
聚物在 3434 cm 处的酰胺 N—H 伸缩振动吸收峰及 由图 3 可见,PASP/3A4HBSA 对 CaCO 3 的阻垢
–1
在 1117 cm 处的酰胺 C—N 振动吸收峰发生了位 率在最初 24 h 内随着处理时间的增加而增加,当处
2+
–1
移,峰的强度明显增强;1597 cm 处羧酸基团的吸 理时间达到 24 h 时,阻垢剂与 Ca 螯合度最大,
收峰发生了比较强的位移;在 1180、1036 和 610 cm –1 PASP/3A4HBSA 的阻垢率为 92.7%,而 PASP 的阻