
Page 40 - 201812
P. 40
·2008· 精细化工 FINE CHEMICALS 第 35 卷
上,用电子扫描显微镜观察其形貌和尺寸;采用 2.2 Fe 3 O 4 @SiO 2 纳米颗粒的 XRD 和 SEM 分析
FTIR 对改性前后 Fe 3O 4@SiO 2 纳米颗粒进行测定,扫 溶剂热法制备 Fe 3 O 4 及其包覆 SiO 2 后所得核壳
1
描次数 32 次,分辨率 4 cm ,波数范围 4000~400 cm 1 结构(Fe 3 O 4 @SiO 2 )纳米材料的结构用 X 射线衍射
采用 Zeta 电位分析仪对改性前后的 Fe 3 O 4 @SiO 2 纳 进行表征,如图 2 所示。
米颗粒悬浮液进行电位测试,测试过程中操作电压
固定为 80 V,频率固定为 3 Hz,计算机进行自动记
录数据,每个样品至少测 3 次,取其平均值;分别对
改性前后的 Fe 3 O 4 @SiO 2 纳米颗粒进行粉体压片,然
后滴 1 g 石蜡油于其表面,稳定 10 min 后用接触角
测量仪测量;用激光共聚焦显微镜对不同罗丹明 B
改性后的 Fe 3 O 4 @SiO 2 纳米颗粒稳定的 Pickering 乳
液进行荧光测试;乳液类型通过电导率仪进行确定。
2 结果与讨论
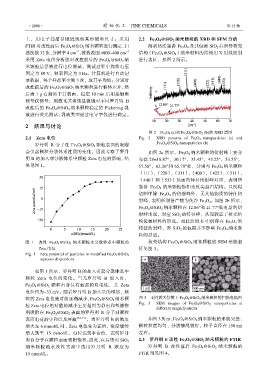
图 2 Fe 3 O 4 (a)和 Fe 3 O 4 @SiO 2 (b)的 XRD 谱图
2.1 Zeta 电位 Fig. 2 XRD patterns of Fe 3 O 4 nanoparticles (a) and
罗丹明 B 分子在 Fe 3 O 4 @SiO 2 颗粒表面的吸附 Fe 3 O 4 @SiO 2 nanoparticles (b)
会引起颗粒分散体系性质的变化,因此考察了罗丹 如图 2a 所示,Fe 3 O 4 纳米颗粒的衍射峰主要分
明 B 的加入对分散体系中颗粒 Zeta 电位的影响,结 布在 2θ=18.87,30.17,35.45,43.25,53.55,
果见图 1。 57.56,62.26和 65.70处,分别与 Fe 3 O 4 纳米颗粒
(111),(220),(311),(400),(422),(511),
(440)和(533)晶面的特征衍射峰对应,表明所
制备 Fe 3 O 4 纳米颗粒物相为反尖晶石结构。且所得
谱图中除 Fe 3 O 4 的衍射峰外,无其他物质的特征衍
射峰,表明所制备产物为纯净 Fe 3 O 4 。如图 2b 所示,
Fe 3 O 4 @SiO 2 纳米颗粒在 12.86和 21.77处有新的衍
射峰出现,对应 SiO 2 的特征峰,从而验证了核壳结
构包覆材料的形成。而且曲线 b 中仍存在 Fe 3 O 4 的
特征衍射峰,即 SiO 2 的包覆并不影响 Fe 3 O 4 纳米颗
粒的晶型。
图 1 改性 Fe 3 O 4 @SiO 2 纳米颗粒水分散体系中颗粒的 核壳结构 Fe 3 O 4 @SiO 2 纳米颗粒的 SEM 形貌表
Zeta 电位 征见图 3。
Fig. 1 Zeta potential of particles in modified Fe 3 O 4 @SiO 2
aqueous dispersions
如图 1 所示,罗丹明 B 的加入引起分散体系中
颗粒 Zeta 电位的变化。当无罗丹明 B 加入时,
Fe 3 O 4 @SiO 2 颗粒自身具有较强的负电性,其 Zeta
电位约为–33 mV。随着罗丹明 B 加入量的增加,颗
粒的 Zeta 电位绝对值逐渐减小,Fe 3 O 4 @SiO 2 纳米颗 图 3 不同放大倍数下 Fe 3 O 4 @SiO 2 纳米颗粒的扫描电镜图
Fig. 3 SEM images of Fe 3 O 4 @SiO 2 nanoparticles at
粒 Zeta 电位绝对值的减小主要是因为静电和氢键作
different magnifications
用吸附在 Fe 3 O 4 @SiO 2 表面的罗丹明 B 分子对颗粒
表面电荷的中和以及屏蔽 [16-17] 。当罗丹明 B 的浓度 如图 3 所示,Fe 3 O 4 @SiO 2 纳米颗粒的形貌完整、
增大至 6 mmol/L 时,Zeta 电位变为正值,浓度继续 颗粒粒度均匀、分散情况较好,粒子直径在 150 nm
增大到至 15 mmol/L,电位达到平台值,表明罗丹 左右。
2.3 罗丹明 B 改性 Fe 3 O 4 @SiO 2 纳米颗粒的 FTIR
明 B 分子在颗粒表面吸附饱和。因此,在后续对 SiO 2
纳米颗粒疏水改性实验中选用罗丹明 B 浓度为 罗丹明 B 改性前后 Fe 3 O 4 @SiO 2 纳米颗粒的
15 mmol/L。 FTIR 图见图 4。