
Page 20 - 201811
P. 20
·1806· 精细化工 FINE CHEMICALS 第 35 卷
中,还原底物Ⅰd 可以进一步与乙腈在氢氧化钾的 及电子云重叠积分,能隙越小、电子云密度质心距
作用下发生迈克尔加成,因此在该反应中需要控制 离越大、电子云重叠积分越小,化合物的 ICT 作用
反应时间在 15 min 内,反应完后立即将反应液倒入 越强,这是还原底物的三苯胺基团给电子效应和含
冰水溶液中淬灭,Ⅰd 最终收率达到 75%。 氮基团拉电子效应共同作用的结果。由图 1 和表 1
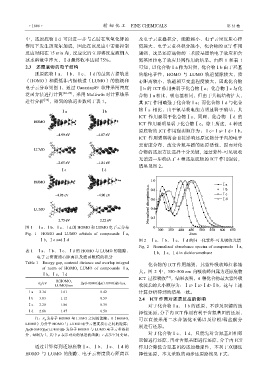
2.3 还原底物的电子结构 可知,以化合物Ⅰa 作为对照,化合物Ⅰb 由于羟基
还原底物Ⅰa、Ⅰb、Ⅰc、Ⅰd 的最高占据轨道 的给电子性,HOMO 与 LUMO 轨道能隙较大,质
(HOMO)和最低非占据轨道(LUMO)的能级和 心距离较小,轨道相互交盖程度较大,因此化合物
电子云分布见图 1,通过 Gaussian09 软件采用密度 Ⅰb 的 ICT 作用要弱于化合物Ⅰa;化合物Ⅰc 与化
泛函方法进行计算 [26-29] ,采用 Multiwfn 对计算结果 合物Ⅰa 相比,吸电基相同,但由于共轭结构扩大,
进行分析 [30] ,得到的轨道参数列于表 1。 其 ICT 作用略强于化合物Ⅰa;而化合物Ⅰd 与化合
物Ⅰa 相比,由于氰基吸电能力明显弱于硝基,其
ICT 作用要弱于化合物Ⅰa,同理,化合物 Ⅰd 的
ICT 作用要明显弱于化合物Ⅰc。综上所述,4 种还
原底物的 ICT 作用强弱顺序为:Ⅰc>Ⅰa>Ⅰd>Ⅰb。
ICT 作用强弱将会直接影响还原底物分子内的电子
云密度分布,改变含氮基团的还原活性,因而对化
合物的还原方法选择十分关键,通过紫外-可见吸收
光谱进一步确认了 4 种还原底物的 ICT 作用强弱,
结果见图 2。
图 1 Ⅰa、Ⅰb、Ⅰc、Ⅰd 的 HOMO 和 LUMO 电子云分布
Fig. 1 HOMO and LUMO orbitals of compounds Ⅰa,
Ⅰb, Ⅰc andⅠd 图 2 Ⅰa、Ⅰb、Ⅰc、Ⅰd 的归一化紫外-可见吸收光谱
Fig. 2 Normalized absorbance spectra of compounds Ⅰa,
表 1 Ⅰa、Ⅰb、Ⅰc、Ⅰd 的 HOMO 与 LUMO 的能隙、 Ⅰb, Ⅰc, Ⅰd in dichloromethane
电子云密度质心距离以及波函数模的积分
Table 1 Energy gap, centroid distance and overlap integral 化合物的 ICT 作用越强,其紫外吸收峰红移越
of norm of HOMO, LUMO of compounds Ⅰa,
大。图 2 中,350~500 nm 内吸收峰归属为还原底物
Ⅰb, Ⅰc, Ⅰd
ICT 过程吸收 [31] ,结果表明,4 种化合物最大紫外吸
R(HOMO,
E g/eV ʃ|φ(HOMO)||φ(LUMO)|dr/a.u.
LUMO)/nm 收波长的大小顺序为:Ⅰc>Ⅰa>Ⅰd>Ⅰb,这与上述
Ⅰa 2.34 1.61 0.42 计算分析得到的结果一致。
Ⅰb 3.03 1.12 0.59 2.4 ICT 作用对还原反应的影响
Ⅰc 2.20 1.86 0.39 对于化合物Ⅰa、Ⅰb 的还原,不涉及双键的选
Ⅰd 2.68 1.47 0.50 择性还原,分子内 ICT 作用有利于含氮基团的还原,
注:E g 为分子 HOMO 与 LUMO 之间的能隙;R(HOMO, 可以直接采用二水合氯化亚锡以及锌粉/稀盐酸分
LUMO)为分子 HOMO 与 LUMO 电子云密度质心之间的距离; 别进行还原。
ʃ|φ(HOMO)||φ(LUMO)|dr 为分子 HOMO 与 LUMO 电子云重叠积
分,量纲为 1, 其中 φ 表示对应的轨道波函数;r 表示空间变量。 对于化合物Ⅰc、Ⅰd,只能先对含氮基团相邻
双键进行还原,再对含氮基团进行还原,分子内 ICT
通过计算得到还原底物Ⅰa、Ⅰb、Ⅰc、Ⅰd 的 作用会降低含氮基团的还原稳定性,不利于双键选
HOMO 与 LUMO 的能隙、电子云密度质心距离以 择性还原,本文采取的两步还原路线见下式: